A perfect genetic hit: New gene mutation implicated in rare congenital diarrhea
When the 1-year-old boy arrived from overseas, he was relying on total parenteral nutrition — a way of bypassing the digestive system to provide nutrients and calories completely intravenously — to survive. From the time of his birth, he had experienced unexplainable diarrhea. Answers were desperately needed.
Sequencing his genes in search of clues, neonatologists and collaborators at the Manton Center for Orphan Disease Research at Boston Children’s Hospital identified a new gene mutation responsible for chronic congenital diarrhea — even finding a similar mutation in two other children as well.
Using patient-derived intestinal organoids in the laboratory, the team discovered that the newly-identified gene mutation, WNT2B, appears to stifle intestinal stem cells’ normal function and growth. The findings were published in the American Journal of Human Genetics.
“A little boy, 16 months of age, was transferred to our care from Kuwait — his diagnosis was failure to thrive,” says Pankaj Agrawal, MD, MMSC, senior author on the study, who is an attending neonatologist at Boston Children’s and the medical director of the Manton Center Gene Discovery Core. “Since his birth, he had suffered severe and chronic diarrhea that interrupted his ability to receive nutrition and grow at a normal rate.”
After identifying the mutation and searching an international gene database, they then found another patient with a similar WNT2B mutation at the Children’s Hospital of Philadelphia. The 7-year-old boy from Vietnam was receiving treatment for nutritional deficiencies and stunted growth related to chronic diarrhea that he had experienced since birth.
“It was a perfect genetic ‘hit,’” Agrawal says.
Making mini organs in the lab
Further confirming a link between WNT2B and congenital diarrhea, the boy from Kuwait soon became a big brother to a newborn baby girl with the same WNT2B mutation and the same symptoms.
Armed with the genetic match, neonatologist Amy O’Connell, MD, PhD in the division of newborn medicine, worked with David Breault, MD, PhD, associate chief of endocrinology, to derive intestinal “organoids” from the patients’ tissue samples. Breault, a co-author on the paper, specializes in creating intestinal organoids for research and gene discovery.
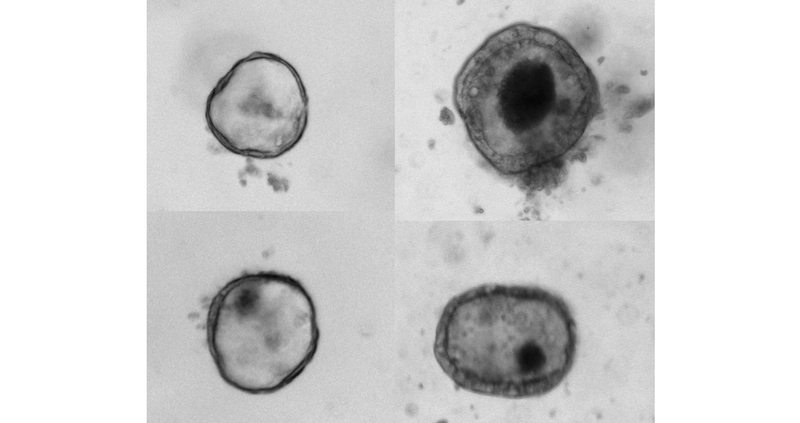
In the lab, O’Connell, who collaborates with Agrawal to functionally evaluate candidate gene mutations, compared the growth of normal intestinal organoids — which typically grow into spherical “miniature organs” containing intestinal stem cell — with organoids containing the WNT2B mutation.
“We found that the organoids with the WNT2B mutation didn’t grow well at all,” says O’Connell, the study’s first author. “These findings obviously have clear implications for the patients involved, but also much wider implications for our understanding of WNT2B’s role in the development of intestinal stem cells.”
They also found that WNT2B-deficient organoids had much lower counts of intestinal stem cells than normal organoids.
“These deficits — fewer intestinal stem cells and diminished ability to support organoid growth — help to explain why the patients with WNT2B mutations experience such severe intestinal disease,” O’Connell says.
In the lab, a disconnect between mice and humans
Interestingly, until now, scientists thought that WNT2B was a redundant gene – that another gene called WNT3A performed the same purpose in normal intestinal growth and development. In earlier mouse experiments, WNT2B mutations didn’t seem to have much impact on overall intestinal health. It was assumed that the same was true in humans. That was before the discovery of WNT2B mutations in the three children, however.
The brother and sister from Kuwait were both seen in Boston Children’s Congenital Enteropathy Program. The program’s co-directors Daniel Kamin, MD, and Jay Thiagarajah, MD, are both co-authors on the paper. While there’s not yet a specific treatment based on the genetic findings, the care team was able to spare the girl from receiving immunosuppressive medications, which had not worked in her older brother and have serious side effects.“Sometimes, the genetics help us not do things,” says Kamin.For more information about the Congenital Enteropathy Program, contact enteropathy@childrens.harvard.edu.
“Our human-derived WNT2B-deficient organoids show that human and mouse models don’t always match,” O’Connell says. “Now we know that at least in humans, WNT2B plays a critical role.”
“There clearly aren’t a lot of these patients, so this observation really came as somewhat of a surprise and contrary to what the scientific literature had documented in mice,” says Breault.
Now, Agrawal says that the team is collaborating on WNT2B mouse models that will hopefully lead them to a therapeutic breakthrough capable of treating patients who have WNT2B mutations.
“For every unique patient that we see, we hunt for a diagnosis and then strive to design therapies that can improve his or her life,” Agrawal says. “Sometimes, for the rarest of the rare cases, it takes us some time to figure out which genes are involved in a unique disorder, but those are the patients that we just can’t stop thinking about until we find an answer for them.”
Related Posts :
-

How Wyatt became the smallest baby to undergo PDA closure at Boston Children’s
In June 2023, at 22 weeks pregnant, Madisen went into labor with twins. Her care team in New Hampshire transferred her to ...
-
Hope in a new home: A family’s journey with HHT
When Yeiden Pérez Camacho imagines the future, he sees himself on a basketball court. At 13, he’s already an ...
-
Beyond average-based medicine: HIE as a blueprint for data-informed care
Historically, outcome prediction in medicine has followed a familiar formula: run a clinical trial, publish the results, guide care based ...
-
The ‘Trach Chapter’: Isabella’s journey with bronchopulmonary dysplasia
Isabella’s life has been anything but ordinary. Born at just 27 weeks gestation and weighing only 1 pound, 4 ounces, Isabella has ...