Overriding resistance to epigenetic inhibitors in neuroblastoma
Neuroblastoma and other children’s cancers pose unique challenges. They’re not caused by the same kinds of genetic mutations that cause adult cancers, and only a minority of their mutations can be targeted with drugs. In a recent study, a team led by Kimberly Stegmaier, MD, at Dana-Farber/Boston Children’s Cancer and Blood Disorders Center systematically deleted every gene in the genome in a number of childhood cancers. This revealed previously unknown, targetable genes that help drive tumor growth.
But Stegmaier is also interested in epigenetic regulators, proteins that influence the regulation of genes and contribute to many pediatric cancers. Since child cancers tend to arise in developing tissues, and epigenetic regulators are active during early development, they’re a hot subject of research. Clinical trials are starting to test drugs that inhibit epigenetic cancer-promoting factors.
There’s a problem, though. Cancers often become resistant to epigenetic inhibitors, as they do with other targeted inhibitors.
“Cancers contain heterogeneous populations of cells, so often one sub-clone will have a way of getting around the drug,” explains Stegmaier, who is also affiliated with the Broad Institute.
Again using genome-wide approaches, she set out to find ways to overcome this resistance. In a study published last December in Cancer Cell, she focused on BET inhibitors and neuroblastoma, the most common cancer in infants and toddlers.
Screening the genome for neuroblastoma drug resistance factors
While BET inhibitors have performed well in prior studies, Stegmaier wanted to be able to predict whether a given patient’s neuroblastoma would become resistant to them.
“We kind of threw the works at this,” she says.

In one approach, her team used what’s known as a genome-wide open reading frame (ORF) screen. Starting with neuroblastoma cells that were sensitive to BET inhibitors, they turned on different genes, one gene per cell, working through the entire genome.
Then they treated the cells with the BET inhibitor JQ1, and watched to see whether boosting any particular gene made the cells resistant to the drug. If so, cells carrying that gene would soon start to predominate; others would die off.
This led to the gene PI3K. When it was boosted, the neuroblastoma cells were unharmed by JQ1.
The researchers then did the opposite experiment. Instead of boosting genes, they used CRISPR gene editing to delete each gene one by one. Their finding: when the gene PTEN was deleted, neuroblastoma cells were resistant to BET inhibitors.
“PTEN suppresses the PI3K pathway,” says Stegmaier, “so both screens were pointing to PI3K.”
All roads lead to PI3K
But her team wasn’t done. They next wanted to see if the same thing happens in nature. And in fact it did. They identified neuroblastoma cells that are naturally resistant to BET inhibitors, and found that PTEN was repressed and PI3K highly activated in these cells, just as in their CRISPR experiments.
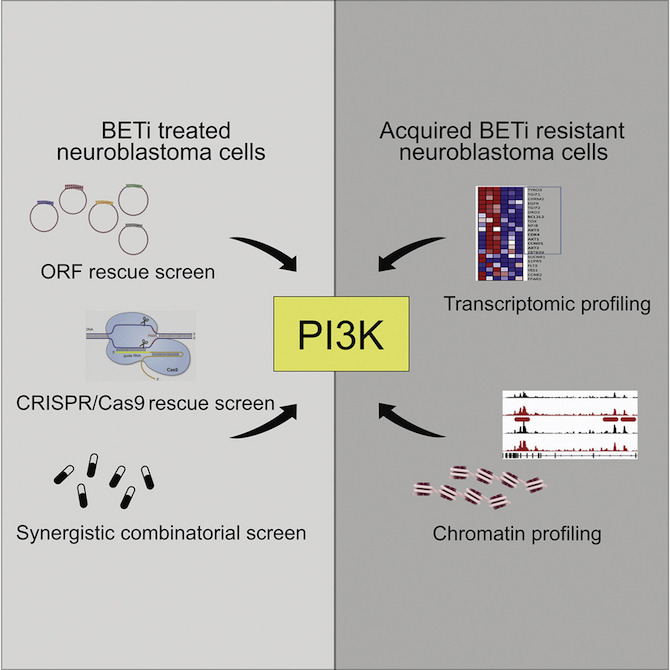
Going further, they took cells that are naturally sensitive to BET inhibitors, and gradually exposed them to increasing amounts of a BET inhibitor until they acquired resistance.
“We were able to tolerize some cell lines over time if we slowly increased the concentration of the BET inhibitor,” Stegmaier explains.
The investigators conducted protein, gene-expression and chromatin profiling of these “persister” cells, and found that the PI3K gene was highly activated. So were other genes called receptor tyrosine kinases (RTKs), which in turn activate PI3K.
“These persister cells were gaining new gene regulatory enhancers, leading to the abnormal activation of the PI3K pathway,” elaborates Stegmaier. “They changed the wiring of their epigenetic state. Our work points to the rewiring of the epigenome, and of enhancers specifically, as another mechanism that contributes to drug resistance.”
BET inhibitors and PI3K inhibitors: a one-two punch?
Finally, putting all of these observations together, the investigators combined BET inhibitors with PI3K inhibitors.
“There was a nice synergy in vitro and in mouse models of neuroblastoma,” Stegmaier says. “We think that this could be a very interesting drug combination moving forward.”
Clinical trials are now testing both BET inhibitors and PI3K inhibitors, but until now, BET inhibitors have only been tested in adults. A newly opened clinical trial, led by Stegmaier’s colleague Steven DuBois, MD, MS, will test the BET inhibitor BMS-986158 in children as young as 1 year with a variety of pediatric cancers.
Since it takes time to get pediatric cancer trials started, these screening approaches can give you a head start in choosing the right compounds.
Stegmaier believes the one-two combination could also be useful in other childhood and adult cancers. In the meantime, she is applying her screening techniques to other pediatric cancers, such as refractory leukemia and Ewing sarcoma.
“Since it takes time to get pediatric cancer trials started, these screening approaches can give you a head start in choosing the right compounds for the second- and third-generation trials,” she says. “That’s very powerful.”
Amanda Balboni Iniguez, PhD, of Dana-Farber/Boston Children’s and the Broad Institute, was first author on the paper. The study was supported by the National Institutes of Health, the NIH intramural research program (NCATS), a Damon-Runyon Fellowship, the Associazione Italiana per la Ricerca sul Cancro-AIRC, a DAAD (Deutscher Akademischer Austauschdienst) fellowship, Hyundai Hope On Wheels, the St. Baldrick’s Foundation and Friends for Life.
Related Posts :
-

How the stars aligned for CeCe’s spinal cord tumor care
Last spring, 6-year-old CeCe was on vacation in Florida when she told her parents, Mike and Mary, that her neck ...
-
Nanoparticle drug combo treats venous malformations
Venous malformations are abnormally shaped veins that develop when the cells lining the blood vessels grow too fast when they ...
-
Mapping ‘neighborhoods’ in aggressive childhood brain tumors
The third-most common kind of childhood brain tumor, supratentorial ependymoma, is an aggressive cancer that often returns after surgery and ...
-
‘Challenge accepted’: Sophia takes on a brain tumor
In 2023, Sophia Mordini landed the role of a lifetime. A competitive dancer, the 12-year-old would play Clara in her company’...