CRISPR-Cas9 screen opens new targets for Ewing sarcoma, other childhood cancers

While the genetic mutations driving adult cancers can sometimes be targeted with drugs, most pediatric cancers lack good targets. That’s because their driving genetic alterations often create fusion proteins that aren’t easy for drugs to attack.
“This is one reason why it is notoriously hard to make targeted drugs against childhood cancers — their cancer-promoting proteins often lack good pockets for drugs to bind to,” says Kimberly Stegmaier, MD.
However, that’s beginning to change. Stegmaier and her colleagues at Dana-Farber/Boston Children’s Cancer and Blood Disorders Center are successfully employing a powerful combination of new genomic and chemical technologies to seek out promising novel targets.
Their collaboration has produced a new clinical trial for patients with Ewing sarcoma and selected other childhood cancers. These cancers typically have one genetic characteristic not shared with most adult cancers — normal, unmutated copies of the TP53 tumor suppressor gene.
Sometimes called the “guardian of the genome,” TP53 normally can help to pull the plug on cells that have turned cancerous. As in the overwhelming majority of pediatric cancers, it usually remains unmutated in Ewing sarcoma, a pediatric bone cancer whose standard treatment includes tough rounds of chemotherapy. Yet, molecular mechanisms somehow are preventing TP53 from doing its job as guardian.
Genomic knockout punch
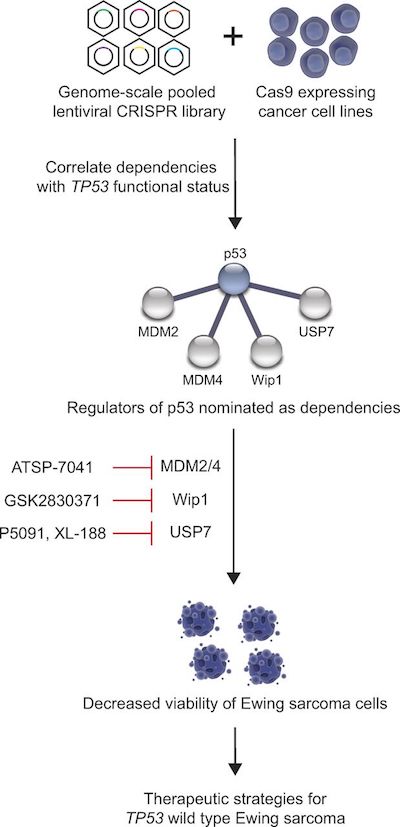
Ewing sarcoma is one of the first pediatric diseases to be put through a screening pipeline that seeks to pinpoint genetic weaknesses for hundreds of types of cancer, through an ongoing project by the Broad Institute, Dana-Farber, Boston Children’s Hospital and Brigham and Women’s Hospital. Using CRISPR-Cas9 gene editing in cancer cell lines, investigators are systematically deleting every gene in the genome, one by one, to see which of these deletions affect cell proliferation.
We’re addressing questions that couldn’t be addressed before, with new genomic technologies and new chemistry.
Most research cell lines for Ewing sarcoma and many other cancers possess a mutated TP53 gene, likely because these cells tend to grow better in the laboratory. But in this case, the researchers wanted to analyze Ewing sarcoma cell lines with normal, “wild-type” TP53, since those are more representative of the disease when it occurs in children and adolescents.
Several gene deletions curbed cell proliferation only in cell lines with the normal TP53 gene, not those with the mutated gene. The team focused on four genes whose proteins can be inhibited by agents that are already in clinical or preclinical evaluation. They validated their findings by testing the agents in cells grown in petri dishes in the lab and in mouse models, and published their results last summer in the Journal of Experimental Medicine.
Targeting the TP53 pathway
Two genes that emerged from the genomic screen were a well-known pair of TP53 regulators called MDM2 and MDM4. In the Ewing sarcoma cell lines, the M2M2 and MDM4 proteins were only active in cells with wild-type TP53. They were also active in mouse models of the tumor.
Stegmaier was already studying the pair for their roles in pediatric leukemia with chemical biologist Loren Walensky, MD, PhD, also at Dana-Farber/Boston Children’s. Years earlier, the Walensky lab had shown that hitting both proteins simultaneously may be an effective way of killing cancer cells, and developed novel “stapled peptide” agents for this two-fisted attack.
One of Walensky’s agents achieved striking results, killing Ewing sarcoma cell lines and slowing tumor growth in mouse models of Ewing sarcoma with wild-type TP53. In one mouse implanted with a patient tumor, the cancer even appeared to vanish completely with treatment.
When clinical trial expert Steven DuBois, MD, heard about these results in a Stegmaier lab meeting, he immediately asked, “When are we going to do the trial?” The clinical study of a stapled-peptide MDM2/MDM4 drug candidate opened this fall.
“It’s super-exciting,” says Stegmaier, thrilled that she and her colleagues could turn basic science discoveries into a pediatric trial within two years.
In pre-clinical studies, she says, the stapled-peptide agent was synergistic with standard chemotherapy for Ewing sarcoma. If the clinical trial of the MDM2/MDM4 experimental drug is successful, the next step might be to combine it with chemotherapy drugs used to treat children who are diagnosed with metastatic disease or who relapse after treatment.
Pre-clinical progress
The CRISPR-Cas9 genomic analysis also opened up avenues to pursue two other genetic targets in the TP53 biological pathway — genes called USP7 and PPM1D.
Though these targets are less established in drug development than MMD2 and MMD4, this effort also pulled in quick help from other Dana-Farber scientists, most dramatically with USP7.
Genomic analyses kept showing USP7 as a strong contender for targeting, but compounds designed to inhibit the USP7 protein initially didn’t perform as expected. That puzzled Stegmaier and her co-workers, until they came across a paper on other more specific USP7 inhibitors developed by the lab of Dana-Farber chemical biologist Sara Buhrlage, PhD.
“We picked up the phone and called her, and she got the compound to us within a day,” Stegmaier says. Experiments with this USP7 inhibitor soon confirmed the genomic evidence and helped to accelerate drug development around the target.
Combination therapy?
Stegmaier’s team also tried combining the agents suggested by the genomic screen, with notable results in the cancer cell lines.
“If you combine the MDM2 and MDM4 inhibitors with USP7 inhibitors, there’s an incredible synergy,” she says. “We’re still very early in thinking about that, but the data suggests that if you could hit this pathway hard, you could kill the cells really effectively. It is our hope that someday these types of targeted therapies might replace cytotoxic chemotherapy.”
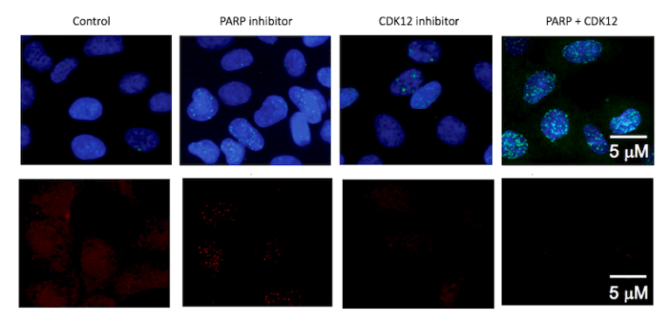
“We’re pursuing novel biology and addressing questions that couldn’t be addressed before, with new genomic technologies and new chemistry,” Stegmaier sums up. “We also think this research will be broadly relevant to other pediatric cancers with normal TP53.”
Björn Stolte of Dana-Farber/Boston Children’s was first author on the paper, and Loren Walensky was co-senior author with Stegmaier. Sara Buhrlage is a co-author. The study was supported by a Hyundai Hope On Wheels grant, the National Institutes of Health, the St. Baldrick’s Foundation and DAAD (Deutscher Akademischer Austauschdienst).
Related Posts :
-

Mapping ‘neighborhoods’ in aggressive childhood brain tumors
The third-most common kind of childhood brain tumor, supratentorial ependymoma, is an aggressive cancer that often returns after surgery and ...
-
A true hero’s journey: How a team approach helped Wolfie overcome pancreatitis
Wolfgang, affectionately known as “Wolfie,” is a bright and energetic 7-year-old with a quick wit and a love for making ...
-
A toast to BRD4: How acidity changes the immune response
It started with wine. Or more precisely, a conversation about it. "My colleagues and I were talking about how some ...
-
A new druggable cancer target: RNA-binding proteins on the cell surface
In 2021, research led by Ryan Flynn, MD, PhD, and his mentor, Nobel laureate Carolyn Bertozzi, PhD, opened a new chapter ...