Why do so many dementia treatments fail? Questioning mouse models of tau accumulation

To date, the search for effective treatments for dementia has yielded only disappointments. Many recent drug candidates target the tau protein, which aggregates and forms tangles in patients’ brain tissue and is involved in 75 percent of all dementias. While tau-targeting drugs have looked promising in mouse models, they’ve failed in clinical trials.
A recent study led by Kathrin Wenger, a PhD student in the lab of Judith Steen, PhD, at Boston Children’s, suggests one reason: The available tau-based mouse models don’t correspond well with tau pathology in late-stage, symptomatic human dementia.
“We don’t think these mice are the right models,” Wenger says.
Tracking tau dynamics
Previous research in the Steen Lab, part of the F.M. Kirby Neurobiology Center, found that the tau protein changes during the course of Alzheimer’s disease as it gets chemically modified. In the new study, Wenger and colleagues analyzed tau in its aggregated form in brain samples from the two most common mouse models. Known as P301S and P301L, these models are based on genetic mutations of tau identified in patients with frontotemporal dementia.
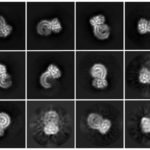
In collaboration with the Proteomics Center at Boston Children’s, Wenger systematically mapped modifications of tau protein at different disease stages, then compared the findings to tau samples from people with Alzheimer’s disease and people with dementia and P301L mutations.
“We took an unbiased approach with quantitative proteomics, looking at all possible chemical modifications of tau over time, and quantified them,” says Wenger. “We are the only group with the technology to quantify modifications comprehensively across a given protein. Other groups focus on one or two chemical modifications previously suspected to played a role in disease, and are not able to quantify these modifications.”
Through meticulous experiments, the Steen team showed that, in both mouse models, the chemical process of phosphorylation drives tau accumulation. The same was true in people with early stage Alzheimer’s disease or dementia with P301L tau mutations. However, in symptomatic human disease and late-stage human Alzheimer’s disease, ubiquitination and acetylation of tau are important, and these were not represented in the mouse models.
Better dementia models needed
The team concludes that the mouse models may be suitable for testing drugs meant to intervene in the early, pre-symptomatic stages of dementia, when phosphorylation is the main tau modification, but not in symptomatic or later-stage disease.
Steen also notes that better models are needed to account for factors such as lifestyle, genetics, environment, comorbidities such as diabetes, and infections that can contribute to human dementia — particularly non-familial Alzheimer’s disease, which makes up more than 90 percent of all Alzheimer’s disease cases.
“We grow mice in optimal conditions, with healthy diets and no stressors or infections,” Steen says. “But human Alzheimer’s disease is the product of multiple biological insults over a lifetime. We need a model that reflects insults people sustain over a lifetime.”
Visit the F.M. Kirby Neurobiology Center at Boston Children’s.
Related Posts :
-

A new angle on the cause of Alzheimer’s disease: Accumulating brain mutations
Alzheimer’s disease is marked by a loss of functional neurons in the brain. But what causes this loss? A ...
-
Tau protein changes correlate with Alzheimer’s disease dementia stage
Research into Alzheimer’s disease has long focused on understanding the role of two key proteins, beta amyloid and the ...
-
Targeting synapse loss in Alzheimer’s to preserve cognition — before plaques appear
Currently, there are five FDA-approved drugs for Alzheimer’s disease, but these only boost cognition temporarily and don’t ...
-
First sharp images reveal structure of key inflammatory protein
After decades of attempts by the scientific community, researchers have now provided the first clear look at a protein implicated ...