

New drug pathway linked with tuberous sclerosis
Tuberous sclerosis complex (TSC) is a neurological disorder causing non-cancerous tumors, called cortical tubers, to grow throughout the brain and body, as well as other conditions like epilepsy and autism. While medications are used to treat some of the manifestations of the disease, safe and more effective treatments targeting disease at a fundamental level are lacking.
Key takeaways:
No drugs are available to treat all manifestations of TSC.
Increased mTOR activity contributes to TSC symptoms.
The heat shock protein (hsp) cascade restores mTOR activity in TSC neurons.
The Hsp pathway represents a potentially druggable target in TSC.
New research from the laboratory of Mustafa Sahin, MD, PhD, hopes to change that. In a new paper published today in Cell Reports, his research team discovered that a cell signaling pathway called the heat shock protein cascade may offer new drugs for TSC.
TSC is caused by mutations in either the TSC1 or TSC2 genes, which together make proteins known as the TSC1/2 protein complex. This protein complex acts on an important complex called the mechanistic target of rapamycin complex 1 (mTORC1). When the TSC1/2 protein complex fails to inhibit mTORC1, the overall mTOR pathway goes into hyperdrive, causing abnormal cell growth and other neurological manifestations of the disease.

In this paper, Sahin’s team showed that the heat shock protein signaling machinery restored normal mTOR activity.
“Finding an alternative pathway, like the heat shock protein pathway, that corrects faulty mTORC1 signaling, may provide new drug targets and expand therapeutic landscape for TSC,” says Sahin, director of the Translational Neuroscience Center and the Translational Research Program at Boston Children’s Hospital.
Too much mTOR, no cilia

Cilia are membrane extensions of a cell’s surface. Some CNS disorders, like brain malformation, autism, and intellectual disability, are known to have mutations in cilia genes and reduced cilia. TSC cells also have less cilia.
Sahin’s team wanted to know more about the potential relationship between cilia and disrupted mTOR activity in neurons. “We wanted to see the crosstalk between these two and see how it was regulated,” says first author Alessia Di Nardo, PhD, research fellow in the Sahin laboratory.
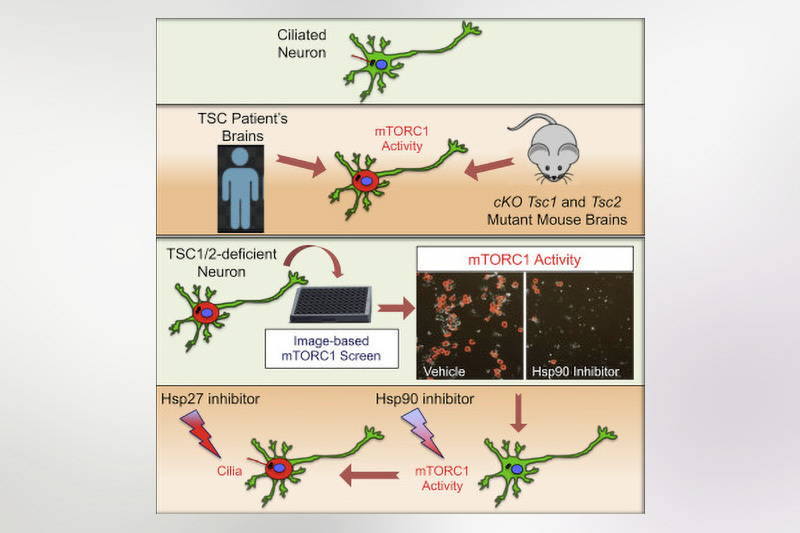
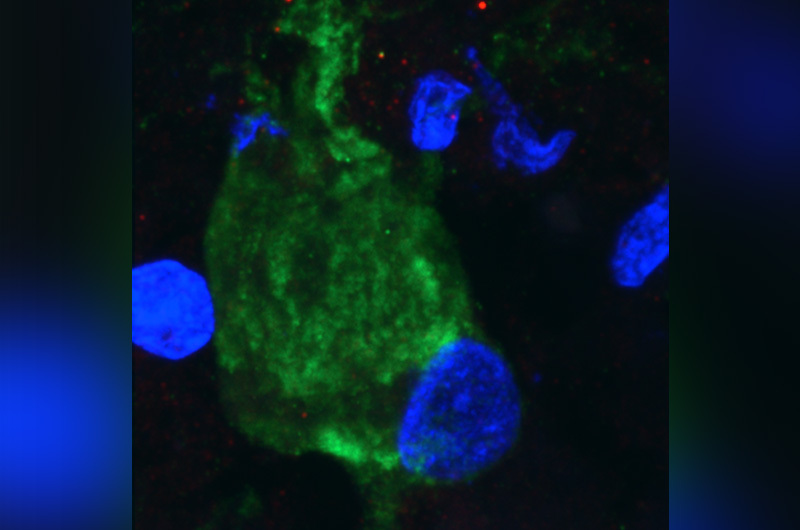
In a mouse model of TSC, they found that the loss of TSC1/2 protein activity in neurons leads to a reduction of cilia. They found the same result studying the giant cells present in cortical tubers brain specimens from TSC patients with epilepsy.
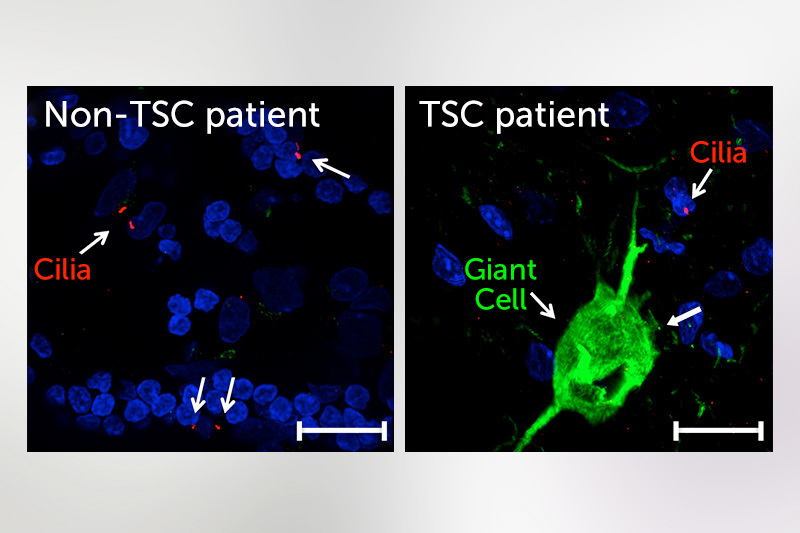
“It is becoming more and more clear that there are a number of neuropsychiatric disorders that have altered cilia, and TSC is among those,” says Sahin. “This puts the cilia as a potentially novel and possibly druggable signaling pathway that can be used to target some of the brain manifestation of TSC.”
Screening reveals a surprise
To identify some of those potential targets, the team set up a drug screening assay. Using mutated rat neuronal cells that lacked TSC1/2, they looked for compounds that interfered with mTORC1 activity causing loss of cilia.
“Our top hit was rapamycin, which confirmed that the screen was robust,” says Di Nardo. The next hits included two inhibitors of Hsp90: geldanamycin (GA) and 17-allylamino-geldanamycin (17-AGG). 17-AGG restored cilia in the rat neuronal cells.

Rat TSC1/2 mutant rat neurons display few cilia (left). Cilia are restored in rat TSC1/2 mutant rat neurons treated with 17-AGG, an inhibitor of heat shock protein 90 (right). Neurons are in green, cilia are in red (Image: Alessia Di Nardo, Boston Children’s)
“This points to the heat shock response as a regulator at different nodes within the mTORC1 signaling cascade,” she adds.
Improving on rapalogs
Since 2010, several compounds called rapalogs are FDA-approved for TSC. Rapalogs are compounds that act like the drug rapamycin and are mTOR inhibitors. While rapalogs have some benefit for treating TSC-associated tumors and suppressing some seizures in some patients, they are ineffective for neuropsychiatric symptoms. And, they can have unwanted side effects.
Sahin’s lab has been trying to identify alternative treatments for TSC that might be potentially more effective, and also maybe even safer than the rapalogs.
HSP 90 has been used as a target for cancer development but has not previously been shown as a target in TSC neurons. The team will now test drugs that inhibit Hsp90 in neuronal mouse models of TSC. Looking forward, they envision using this screening platform for identifying other potential drugs for TSC-related neuronal cell dysfunction.
This paper is the latest in TSC research at Boston Children’s. Since October 2019, Sahin has been director of the Developmental Synaptopathies Consortium (DSC), a 10-center NIH-funded research program established to better understand the pathophysiology of TSC and two other rare genetic disorders, and to discover new treatments.
Other authors from Boston Children’s include Isadora Lenoël, Kellen Winden, Alina Rühmkorf, Meera Modi, Lee Barrett, Ebru Ercan-Herbst, Pooja Venugopal, Robert Behne, and Robin J. Kleiman. Carla A.M. Lopes and Mónica Bettencourt-Dias from Instituto Gulbenkian de Ciência, Portugal are co-authors.
Learn more about the Tuberous Sclerosis Program at Boston Children’s.
Related Posts :
-

A case for Kennedy — and for rapid genomic testing in every NICU
Kennedy was born in August 2025 after what her parents, John and Diana, describe as an uneventful pregnancy. Soon after delivery, ...
-
The journey to a treatment for hereditary spastic paraplegia
In 2016, Darius Ebrahimi-Fakhari, MD, PhD, then a neurology fellow at Boston Children’s Hospital, met two little girls with spasticity ...
-
Thanks to Carter and his family, people are talking about spastic paraplegia
Nine-year-old Carter may be the most devoted — and popular — sports fan in his Connecticut town. “He loves all sports,” ...
-
Genomic sequencing transforms a life: Asa’s story
Asa Cibelli feels like he’s been reborn. The straight-A middle schooler plays basketball and football, does jiu jitsu, is ...



