

Shooting for the moon: From diagnosis to custom drug, in one year

Ed. note: Mila passed away in February 2021, at age 10. The Mila’s Miracle Foundation continues to work to pave a pathway for personalized treatments. The FDA recently released a draft guidance on testing custom antisense oligonucleotides (ASOs) in patients.
One weekend in January 2017, Timothy Yu, MD, PhD, was relaxing at home when his wife said, “Hey Tim, will you look at this post on Facebook? This might be something you’d be interested in.”
Doctors had just diagnosed a young girl named Mila in Colorado with Batten disease, a rare and fatal neurodegenerative disorder. After a full clinical genetic workup, they could find only one of her two mutations. They suggested the family simply enjoy their remaining time with Mila. Even worse, without locating Mila’s second mutation, they couldn’t rule out the disease in her younger brother Azlan.
Mila had just turned 6 and was quickly losing her sight, language, and ability to walk. Her mother pleaded: Could someone sequence Mila’s entire genome, and do so quickly?
‘A lot like my own kids’
It was a pivotal moment. Yu, a neurologist and an attending physician in the Division of Genetics and Genomics at Boston Children’s Hospital, happens to run a research lab with deep expertise in whole genome sequencing. The lab’s specialty was finding unusual genetic mutations that cause neurologic disorders.
“Most clinical labs focus on just 1 percent of the genome,” Yu says. “The 99 percent that’s left over, called noncoding DNA, is the dark matter of the genome. Our lab has a particular interest in that, so that’s where we thought we could help.”
Yu read the family’s story on the Mila’s Miracle Foundation website, formed by her mother, Julia Vitarello.
“I saw a young girl who was incredibly active, vivacious, happy,” says Yu. “She loved to sing, she loved to talk. You could tell that she was incredibly social and had a lot of energy. She reminded me a lot of my own kids.”
Finding a needle in a haystack
After a phone conversation, Mila’s family quickly agreed to ship blood samples to Yu in Boston. In February, his team got the genetic results back. Yu and lab member Aubrie Soucy pored over them for a week.
“First, we looked for all the traditional types of mutations — point mutations, copy number variations, and other large deletions or duplications — and found nothing,” says Yu. “We then looked through each of the 14 known Batten disease genes. Still nothing. Ultimately, we decided to sit down and inspect the raw data ourselves.”
After days of manual inspection, they noticed an odd pattern in Mila’s genetic sequence — as well as Julia’s. The reads partially matched the expected sequence in a standard reference genome. But one portion was markedly different. “We realized that a foreign DNA insertion had landed in the middle of Mila’s CLN7 gene,” says Yu. “That insertion was what was breaking the gene.”
In April 2017, Yu phoned Mila’s parents to share the genetic findings. “We tested her little brother, and we were able to tell Mom and Dad that Azlan had no risk of developing this disease,” he says.
Yu had a proposal to share with the family.
“He told us, ‘There’s one more thing. We have an idea of how we might be able to help Mila,” says Julia.
An unwanted guest
“Jumping genes,” known as transposons, are ubiquitous in our genomes. Many scientists believe they play a role in evolution, helping us quickly acquire new traits. But in Mila’s case, as Yu’s team confirmed during May and June 2017, the inserted DNA was an unwanted guest that created what the cell thought was an exon, or protein-coding gene. It was disrupting assembly of the CLN7 gene, in turn compromising production of the CLN7 protein.
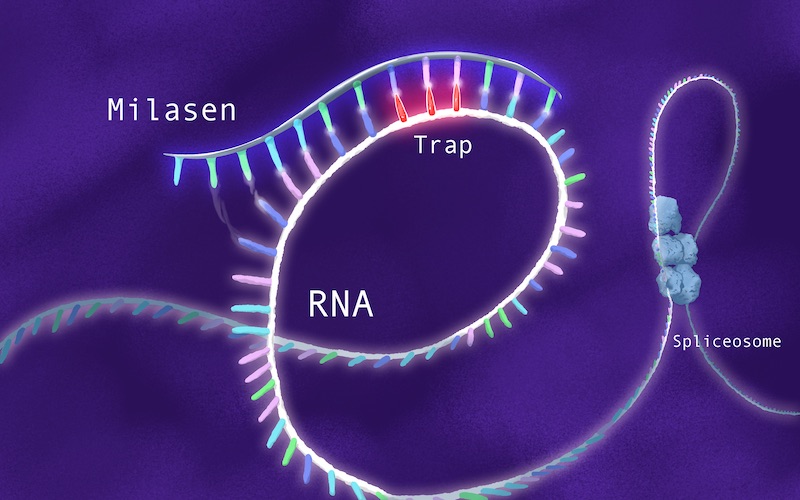
Mila’s transposon is a retrotransposon, meaning it uses the cell’s replication machinery to copy itself. Fortuitously, Alice Lee, PhD, a computational biologist, had recently joined the Division of Genetics and Genomics. One of the world’s experts on retrotransposons, Lee had developed algorithms for detecting them in whole genome sequencing data.
“The fact that Mila’s mutation was something that we actually knew something about was a real stroke of luck,” says Yu.
Standing on Spinraza’s shoulders
The Food and Drug Administration (FDA) had recently approved a drug called nusinersen (Spinraza) for spinal muscular atrophy, a disease in which the motor neurons waste away, causing muscles to weaken. Spinraza is what’s known as an antisense oligonucleotide: a segment of artificial genetic code designed to mirror a piece of code in the genome, bind to it, and change how the genome is read. In spinal muscular atrophy, it boosts a “backup” gene, SMN2, so it can fully stand in for the mutated SMN1 gene.
Spinraza has been a rare-disease success story, improving strength and motor function in many children and sparing many from needing to go on ventilators. Boston Children’s was a lead site for the clinical trials.
“Spinraza became an inspiration for what we could try to offer to Mila,” says Yu. “We said, ‘why can’t we take that same approach and customize it for Mila?’”
Although the family’s foundation had been raising money to develop a gene therapy approach to treatment, the oligonucleotide approach intrigued Yu. Data suggest that oligonucleotides, given through a spinal injection, get into 90 percent of cells in the spinal cord and brain, much more than gene therapy could likely achieve. And oligonucleotides appeared to be very safe.
In Mila’s case, Yu’s team sought to design an oligonucleotide that would home to the CLN7 gene and hide the unwanted retrotransposon, causing the cellular machinery to skip over it (hence the term “exon skipping”).
“It would be sort of a molecular Band-aid, allowing the gene to assemble properly,” says Yu. “So we started off saying, ‘Let’s try to replicate the drug development path for Spinraza.’”
Proof of concept
During the summer of 2017, Yu began designing a series of exon-skipping oligonucleotides, and lab members April Hu, MD, PhD; Aubrie Soucy; Jai Vaze; Renata Didonato; and Jinkuk Kim, PhD, began testing them. In early fall, they showed that three candidate drugs, introduced into Mila’s skin cells in a dish, correctly targeted her mutation and corrected the faulty splicing. They also restored healthy function of Mila’s lysosomes — cellular “recycling centers” that had become overwhelmed, unable to empty their trash.
Each of the 14 known genes associated with Batten disease appears to be involved in some aspect of lysosomal function. When lysosomes malfunction, the cell tries to compensate by making more lysosomes, which also fill up and eventually overflow. Yu’s team, aided by the TransLab at Boston Children’s and Joe Mazzulli, PhD, at Northwestern, showed that when oligonucleotides were added to Mila’s cells, the number of lysosomes was reduced, as was the waste spilling out into the cell.
Yu and his colleagues named their top candidate “milasen.” But they were still far from having an actual treatment. They needed partners to produce the drug in quantity, under proper manufacturing conditions, and help test it for safety and bioavailability.
And they needed to convince the FDA to approve a clinical “n of 1” trial — testing milasen just in Mila, and without first showing its efficacy in animal models of Batten disease. Because in fact, no animal models of her mutation existed.
Industry allies
Yu began calling around to try to find industry partners. His calls eventually led him to TriLink BioTechnologies and Brammer Bio, which helped manufacture milasen on an expedited timeline. For toxicology help, he reached out to Charles River Laboratories. He was referred to Lauren Black, PhD, a senior scientific consultant, who also became a key advisor.

“Prior to joining Charles River, Lauren was a toxicologist at the FDA,” says Yu. “She became an expert on all the different ways that early-generation oligonucleotide drugs could go wrong, and had written many of the FDA guidances about how to safely approach oligonucleotides in humans. She was the perfect person to advise us.”
In another stroke of luck, Yu managed to wrangle a speaking invitation to DIA/FDA Oligonucleotide-Based Therapeutics Conference — just weeks away in October 2017. The conference, held every two years, draws a mix of industry and regulatory professionals.
“We had in the audience exactly the people that would be helping us in the months to come,” says Yu. “They had been working on therapeutic applications of oligonucleotides for decades, but everyone was really interested to see what we had done, and how far this case might actually push us — even for a single child.”
Yu came away with a stack of business cards from additional people willing to volunteer their advice and round out his team, including a former deputy commissioner of the FDA and a medical ethics consultant who had been involved in the FDA approval of Spinraza.
A precedent-setting case
Separately, Julia and Mila’s Miracle Foundation had been racing to raise money. In November 2017, the stars began to align for Mila to be treated.
“It gradually evolved from a completely theoretical possibility, going from just hearing this word ‘exon skipping’ and slowly moving through all of these steps,” says Mila’s father, Alek. “We finally realized that the foundation had enough money to see this through and that Dr. Yu had enough science to convince the FDA that it might work. Just before Christmas is when we realized that this was going to happen.”
The FDA granted official permission in January 2018. It set a new precedent. The agency had previously allowed existing drugs to be repurposed for seriously ill patients with no other options. But no one had ever used this path to test a brand-new drug created for a single patient.

A test for personalized medicine
Meanwhile, Yu had brought Mila’s case to leaders around Boston Children’s, including Chief Scientific Officer David Williams, MD, PhD; Olaf Bodamer, MD, PhD, associate chief of Genetics and Genomics; and Chief Pharmacy Officer Al Patterson, PharmD. Charles Berde, MD, PhD, an expert in spinal drug delivery, and Alessandra Biffi, MD, director of the Gene Therapy Program, also weighed in, as did the hospital’s bioethics committee.
Eventually, everyone agreed that milasen was worth trying. Mila was fast declining, and her case exemplified the hospital’s commitment to offering personalized treatments for rare conditions.
“This case did bring up lots of unexplored risks for the institution,” Yu acknowledges. “We were moving into a space where not only didn’t we have a lot of experience, no one else did either. But at the end of the day, everyone was convinced by the mission. It became not why, but how we would manage each hurdle.”
Treatment begins
The hospital’s ICCTR (led by Andrew Place, MD, PhD) and Experimental Therapeutics Unit (led by Cindy Williams) provided planning and implementation help. In late January 2018, two weeks after the FDA’s permission, Mila received the first of a series of nine escalating, biweekly doses of milasen, given under general anesthesia. These continued through June 2018. In August 2018, she received her first maintenance treatment, given every three months.

To date, Mila has mostly done well, with no notable adverse effects. Although her clinical decline had continued in the months leading up to the trial, she has shown stabilization in many clinical domains since starting milasen.
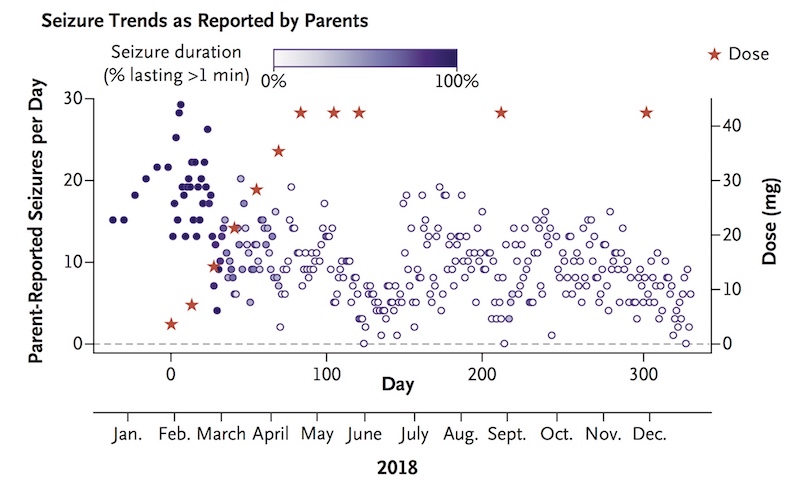
Where she once had about 30 seizures daily, lasting one to two minutes, Mila now typically has zero to 10 seizures daily, usually lasting a few seconds. The team continues to monitor her closely, and in March 2019, decided to increase the frequency of her injections to every two months. (They are now given at Mila’s home hospital in Colorado.) Yu hopes this will further curb her seizures, which seemed to trend up near the end of each three-month period.
Mila’s therapy and her early results were published October 9 in The New England Journal of Medicine. Jinkuk Kim and April Hu are co-first authors on the paper.
Meanwhile, Julia reports that Mila has been mostly stable with some important improvements since starting her treatment last year. Her seizures are barely noticeable now, her swallowing has improved, and she holds her neck and body up much better when standing and walking, still with assistance. At times, she seems to be trying to speak. Her responsiveness to stories and songs isn’t as frequent as last year, but it seems to surge immediately after receiving milasen. Julia remains cautiously optimistic, while continuing to fight alongside her daughter.
The start of a new path
The creation of milasen, in less than a year, could signal the beginning of a revolution in how genetic conditions are treated. Boston Children’s is converting Yu’s process for developing milasen — from diagnosis to synthesis and treatment — into a protocol for handling similar cases at the hospital, one patient at a time. Many other cases could eventually be treatable with oligonucleotides, which can correct splicing errors or boost levels of a gene, Yu believes.
Informed by Yu’s efforts, Boston Children’s is examining the best way to accelerate its discovery science into new therapies for children with rare conditions. The hospital has also developed an Oversight Committee on Personalized Experimental Therapeutics to review similar complex cases.
“There are no commercial incentives to handle these patients right now,” Yu notes. “There are whole categories of diseases with populations too small to attract industry effort. But in a children’s hospital with a major focus on research, we can focus on these rare conditions. I think think that some of the most exciting parts in science are when you try to do something new, when there isn’t a recipe.”
To support Yu’s research, contact Lisa.Slater@chtrust.org
Related Posts :
-

Hope in a new home: A family’s journey with HHT
When Yeiden Pérez Camacho imagines the future, he sees himself on a basketball court. At 13, he’s already an ...
-
Gene therapy for hearing loss: Tag-teaming from the lab to the clinic
Two-year-old Miles is one of the first patients with hereditary hearing loss to receive gene therapy at Boston Children’s ...
-
A case for Kennedy — and for rapid genomic testing in every NICU
Kennedy was born in August 2025 after what her parents, John and Diana, describe as an uneventful pregnancy. Soon after delivery, ...
-
The journey to a treatment for hereditary spastic paraplegia
In 2016, Darius Ebrahimi-Fakhari, MD, PhD, then a neurology fellow at Boston Children’s Hospital, met two little girls with spasticity ...



