

Optimized CRISPR/Cas9 gene editing averts hearing loss in ‘Beethoven’ mice
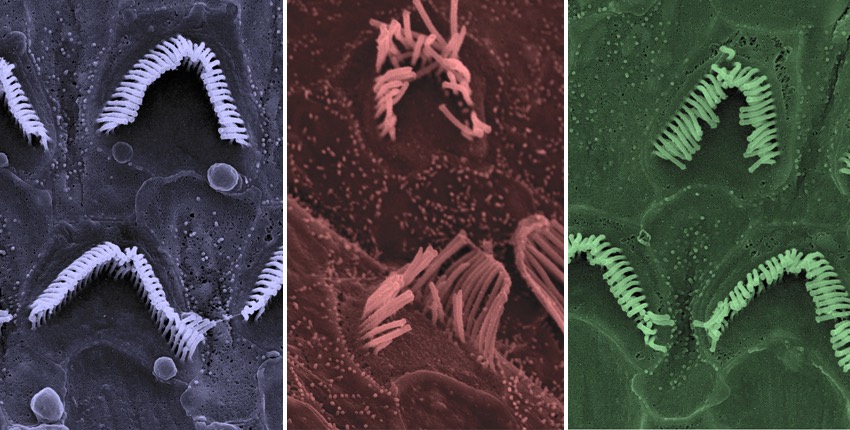
Using a novel gene-editing approach, scientists at Boston Children’s Hospital and Harvard Medical School have salvaged hearing in a mouse model of hereditary deafness, with no apparent off-target effects. The system successfully identified a single misspelled “letter” in the defective copy of a gene required for hearing, disabled this aberrant copy, and spared the healthy one. It could be applicable to other dominantly inherited diseases, the researchers believe.
In 2011, a team led by Jeffrey Holt, PhD, of the F.M. Kirby Neurobiology Center at Boston Children’s Hospital, demonstrated the Tmc1 protein is required for hearing and balance. In 2015, Holt and colleagues restored hearing in “Beethoven” mice with gene therapy, using an engineered adenovirus to deliver a functioning copy of the Tmc1 gene into the ear. (Beethoven mice typically go completely deaf by 6 months of age; people with defects in the same gene become profoundly deaf by their mid-20s.)
The new work, described today in Nature Medicine, used CRISPR-Cas9 gene editing instead of traditional gene therapy, optimizing it to make it safer and more precise.
“We believe our work opens the door toward a hyper-targeted way to treat an array of genetic disorders that arise from one defective copy of a gene,” says Holt, who was co-first author on the study with David Corey, PhD, of Harvard Medical School’s Blavatnik Institute. “This truly is precision medicine.”
This video by Rick Groleau and Ekaterina Pesheva at Harvard Medical School encapsulates what they’ve done:
Refining CRISPR/Cas9 gene editing
So far, classic CRISPR-Cas9 gene editing systems have had less-than-perfect accuracy: The Cas9 enzyme that snips out the mutant DNA sequence can end up cutting the wrong DNA. To add precision, the researchers adapted a tool originally developed by Keith Joung, MD, PhD, and Ben Kleinstiver, PhD, of Harvard Medical School and Massachusetts General Hospital. It uses a modified Cas9 enzyme, derived from Staphylococcus aureus instead of Streptococcus pyogenes, that can pinpoint the specific DNA mutation in Beethoven mice.
The system thus combines two levels of recognition — the modified Cas9 enzyme, and the guide RNA that leads it to the mutant sequence. As a result, it was able to spot a single incorrect DNA letter in the Tmc1 gene among 3 billion letters in the mouse genome (in this case, an “A” swapped in for a “T”).
“Our results demonstrate that this more refined, better targeted version of the now-classic CRISPR/Cas9 editing tool achieves an unprecedented level of identification and accuracy,” says Corey.

In an initial set of experiments in cells with and without the Beethoven mutation, the tool accurately distinguished between mutant DNA and normal DNA in copies of the Tmc1 gene. In Beethoven cells, which contained one defective and one normal copy of Tmc1, at least 99 percent of the molecular “cuts” occurred exclusively in the defective copy.
When researchers injected the gene-editing treatment into the inner ears of mice with and without the Beethoven mutation, editing activity occurred only in mice with the Beethoven defect, as evidenced by DNA analysis. Treated mice without the mutation showed no editing changes in their inner ear cells. When scientists stimulated their hair cells, the cells showed unchanged, normal responses — confirming that the gene-editing therapy had no effect on normal gene function.
Curbing hearing loss
But what about hearing? The researchers performed the gold-standard test: They measured the animals’ auditory brainstem responses, which capture how much sound is detected by hair cells in the inner ear and transmitted to the brain.

Two months after the gene-editing therapy, the Beethoven mice exhibited markedly better hearing than untreated siblings also carrying the Tmc1 mutation. Those treated were able to detect sounds at about 45 decibels, the level of a normal conversation — about 16 times quieter than sounds detected by untreated mice. One treated mouse was capable of hearing sounds at 25 to 30 decibels, a level similar to a whisper, virtually indistinguishable from its healthy peers.
Because the disease is marked by progressive hearing loss, the researchers assessed the effect of therapy over time. After treating some of the mice shortly after birth, they tested hearing levels every four weeks for up to six months.

In month one, the untreated Beethoven mice could hear low-frequency sounds but had notable hearing loss at high frequencies; by month six, they had lost all their hearing. In contrast, treated Beethoven mice retained near-normal hearing at low frequencies, and some showed near-normal hearing even at high frequencies. Even more encouragingly, a small subset of these mice that were followed for nearly a year retained stable, near-normal hearing.
Intact hearing cells
Notably, animals without the genetic defect did not experience hearing loss as a result of the gene therapy — more evidence that the treatment selectively targets the aberrant copy of the gene.
The researchers also used electron microscopy to visualize the hearing cells in the inner ears of the Beethoven mice. As expected, they saw gradual loss of hearing cells in the untreated mice, along with deterioration in their structure. By contrast, both the treated Beethoven mice and the treated healthy mice retained a normal number of hearing cells with intact or near-intact structure.
Finally, the scientists tested their treatment in a line of human cells carrying the Beethoven mutation. As in the mice, DNA analysis showed that editing occurred exclusively in the mutant copy of the Tmc1 gene, sparing the normal one.
Targeting other diseases
Because of its ability to target single-point genetic mutations, the researchers believe the new CRISPR/Cas9 approach holds promise for 15 other genetic forms of inherited deafness also caused by a single-letter mutation. They also believe their technique could be adapted for use in as many as 20 percent of other dominantly inherited genetic diseases caused by single-point mutations.
This treatment could be developed to selectively silence genes that carry single-point mutations and potentially treat many other forms of human disease.
To make this calculation, the scientists scanned a national repository of all known genetic mutations linked to human diseases, known as ClinVar. The analysis showed that based on the tool’s specificity, it could correctly identify 3,759 defective gene variants collectively responsible for one-fifth of dominant human genetic mutations.
“To be sure, this is the first step in a long journey,” Holt said. “But what we have here is proof of principle that demonstrates this highly specific, highly targeted treatment could be developed to selectively silence genes that carry single-point mutations and potentially treat many other forms of human disease.”
Bence György, MD, PhD, of Harvard Medical School, was the paper’s first author. (He is now at the Institute of Molecular and Clinical Ophthalmology in Basel, Switzerland). Holt holds patents on Tcm1 gene therapy and is an advisor to several biotech companies focused on inner-ear therapies. Joung has financial interests in several companies. See the paper for a list of authors, funders, and disclosures.
This story is adapted from a press release from Harvard Medical School.
Related Posts :
-

Hope in a new home: A family’s journey with HHT
When Yeiden Pérez Camacho imagines the future, he sees himself on a basketball court. At 13, he’s already an ...
-
Gene therapy for hearing loss: Tag-teaming from the lab to the clinic
Two-year-old Miles is one of the first patients with hereditary hearing loss to receive gene therapy at Boston Children’s ...
-
A case for Kennedy — and for rapid genomic testing in every NICU
Kennedy was born in August 2025 after what her parents, John and Diana, describe as an uneventful pregnancy. Soon after delivery, ...
-
The journey to a treatment for hereditary spastic paraplegia
In 2016, Darius Ebrahimi-Fakhari, MD, PhD, then a neurology fellow at Boston Children’s Hospital, met two little girls with spasticity ...



