

Could a pill treat sickle cell disease?
The new gene therapies for sickle cell disease — including the gene-editing treatment Casgevy, based on research at Boston Children’s Hospital — have been game-changing for the patients who have received them. But Stuart Orkin, MD, the Boston Children’s hematologist whose work led the way to Casgevy, wants to go even further.
“The editing therapy is a wonderful achievement, but only a small fraction of the people who could benefit will ever receive it,” Orkin says. “If we had a pill that could do what the genetics do, that would be great.”
In new work just published in Science, Orkin and his team at Dana-Farber/Boston Children’s Cancer and Blood Disorder Center have taken an important step in that direction. Their discovery may chart a path to a small molecule drug that could be made available to all patients with sickle cell disease, as well as those with beta thalassemia.
Restarting production of fetal hemoglobin
In sickle cell disease, a gene mutation causes the hemoglobin molecules in red blood cells to form a rigid chain. This causes the cells to stiffen into a sickle shape, clogging blood vessels and causing pain episodes. Casgevy works by enabling people to produce a fetal form of hemoglobin that doesn’t cause sickling. It does so by suppressing the gene BCL11A, which normally shuts down fetal hemoglobin production after birth. This lets fetal hemoglobin production resume.
However, Casgevy and other gene therapies for sickle cell disease are complex, requiring nine months to a year to complete, including chemotherapy and weeks of hospitalization. Orkin’s dream has been to find a small-molecule drug that could be taken orally — bringing treatment to large populations of people.

Early work in the 1980s, in which Orkin participated, identified one oral drug, hydroxyurea, that raises fetal hemoglobin levels. The FDA approved it in 1998. But Orkin says hydroxyurea and other available drugs don’t work in all patients and are not very potent.
“The BCL11A protein is the best and most direct target,” he says, “but it is difficult to hit and has sometimes been called undruggable.”
Destabilizing BCL11A
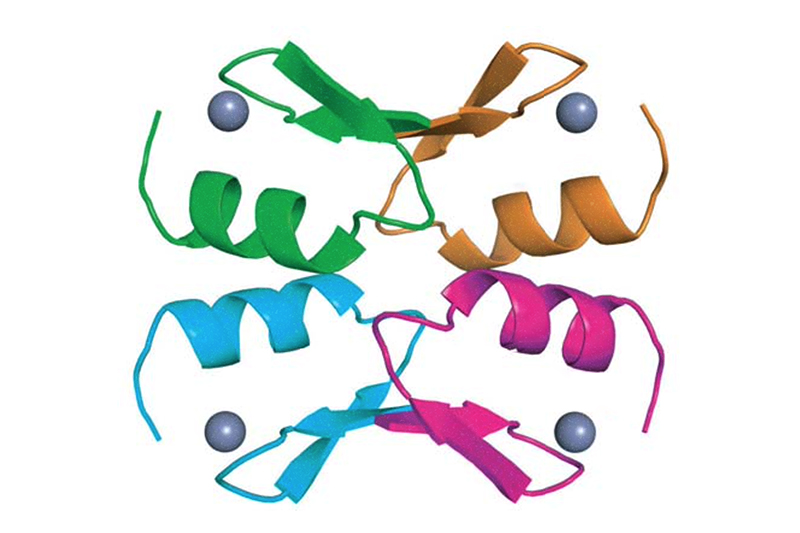
In the new work, Orkin and his colleagues explored the structure and chemistry of the BCL11A protein, looking for ways to hit it. They found that in order for BCL11A to be stable in cells and capable of shutting down fetal hemoglobin production, it must assemble into a tetramer — four connected BCL11A molecules. It does so with the help of a zinc finger, a small protein wrapped around a zinc molecule.
Orkin’s team has now reported the crystal structure of the BCL11A tetramer and suggested several ways to attack it with small molecules.
“The tetramer is like a blob with nooks and crannies,” Orkin says. “The approach we’re working on is to put small molecules into the surface pockets. We think this might destabilize the tetramer, or draw in the cell degradation system to break it down.”
Orkin and his colleagues are further investigating this and other strategies. “It took a long time to put the whole story together,” he says. “I think this will open up new approaches to drug development.”
Ge Zheng, PhD, Maolu Yin, PhD, and Stuti Mehta, PhD, in the Orkin lab were co-first authors on the paper.
Learn more about sickle cell disease and the Gene Therapy Program at Boston Children’s.
Related Posts :
-

Sickle cell gene therapy and boosting fetal hemoglobin: A 75-year history
Ed. Note: This post updates an earlier post from 2018. In a landmark decision today, the Food and Drug Administration (FDA) ...
-
A sickle cell first: Base editing, a new form of gene therapy, leaves Branden feeling ‘more than fine’
Though he doesn’t remember it, Branden Baptiste had his first sickle cell crisis at age 2. Through elementary school, he ...
-
How growing up with sickle cell disease is shaping Nancy’s future
Imagine appearing completely healthy while managing a life-threatening condition. Most people who meet Nancy Blankson would never know she has ...
-
Manny: Hoping new research helps others with sickle cell disease
Emmanuel “Manny” Johnson, Jr., shares many loves with his little brother, Aiden — from basketball to video games. One thing he ...



